
發佈日期: 2020-09-18
[Bruker Newsletter]Bruker引領MALDI-2和4D-蛋白質體學發展,再成ASMS 2020焦點
全新MALDI-2離子源大幅提升小分子分析靈敏度
Bruker一直以MALDI技術聞名全球,並已在質譜影像領域上的應用與創新居全球領導地位;自推出timsTOF Pro創新體學系統後,在深度覆蓋和高通量蛋白質體學分析中達成了突破性的進展,並且迅速和全球研究機構、大學院校和私人企業開展了一系列深度的合作。2019年,Bruker結合蛋白體學和質譜成像的技術優勢,推出了timsTOF fleX,為空間定位體學配置了可切換的ESI和MALDI雙游離源。從timsTOF Pro到timsTOF fleX,Bruker兩項重要產品的發佈都與科學家緊密合作,比如:Matthias Mann、Ruedi Aebersold,Ben Collins和Hannes Roest、Jürgen Cox等;產生了PASEF(Parallel Accumulation Serial Fragmentation平行累積序列碎裂)、diaPASEF(資料非依賴採集的PASEF)、MaxQuant等合作成果,更快地進行蛋白質體學深度覆蓋的研究。深耕更廣泛的臨床研究的市場,例如: 皇家墨爾本醫院的Andrew 教授使用timsTOF Pro,在17分鐘的液相梯度,平均鑑定6,600個蛋白,每天運行50個樣本。日本京都大學的Yasushi教授研究藥物與激酶活性,發展了1分鐘梯度,每天運行864個樣本的方法。
2019年ASMS上推出了timsTOF fleX質譜儀,包括於ESI timsTOF Pro™質譜的軟體可切換的MALDI源,這種新型ESI /MALDI組合功能,可在同一台質譜上進行空間定位體學SpatialOMx™,可在高空間解析度下進行快速、無標記的MALDI成像,而在今年ASMS 2020網路會議上,Bruker推出了世界上第一個商用MALDI後電離(PI)離子源MALDI-2 (圖1),可用於timsTOF fleX質譜儀上,全新的MALDI-2離子源可將傳統對MALDI不敏感的小分子和脂質分析的靈敏度提高一到兩個數量級,廣泛的擴大MALDI技術的質譜成像應用範圍。
Bruker一直以MALDI技術聞名全球,並已在質譜影像領域上的應用與創新居全球領導地位;自推出timsTOF Pro創新體學系統後,在深度覆蓋和高通量蛋白質體學分析中達成了突破性的進展,並且迅速和全球研究機構、大學院校和私人企業開展了一系列深度的合作。2019年,Bruker結合蛋白體學和質譜成像的技術優勢,推出了timsTOF fleX,為空間定位體學配置了可切換的ESI和MALDI雙游離源。從timsTOF Pro到timsTOF fleX,Bruker兩項重要產品的發佈都與科學家緊密合作,比如:Matthias Mann、Ruedi Aebersold,Ben Collins和Hannes Roest、Jürgen Cox等;產生了PASEF(Parallel Accumulation Serial Fragmentation平行累積序列碎裂)、diaPASEF(資料非依賴採集的PASEF)、MaxQuant等合作成果,更快地進行蛋白質體學深度覆蓋的研究。深耕更廣泛的臨床研究的市場,例如: 皇家墨爾本醫院的Andrew 教授使用timsTOF Pro,在17分鐘的液相梯度,平均鑑定6,600個蛋白,每天運行50個樣本。日本京都大學的Yasushi教授研究藥物與激酶活性,發展了1分鐘梯度,每天運行864個樣本的方法。
2019年ASMS上推出了timsTOF fleX質譜儀,包括於ESI timsTOF Pro™質譜的軟體可切換的MALDI源,這種新型ESI /MALDI組合功能,可在同一台質譜上進行空間定位體學SpatialOMx™,可在高空間解析度下進行快速、無標記的MALDI成像,而在今年ASMS 2020網路會議上,Bruker推出了世界上第一個商用MALDI後電離(PI)離子源MALDI-2 (圖1),可用於timsTOF fleX質譜儀上,全新的MALDI-2離子源可將傳統對MALDI不敏感的小分子和脂質分析的靈敏度提高一到兩個數量級,廣泛的擴大MALDI技術的質譜成像應用範圍。
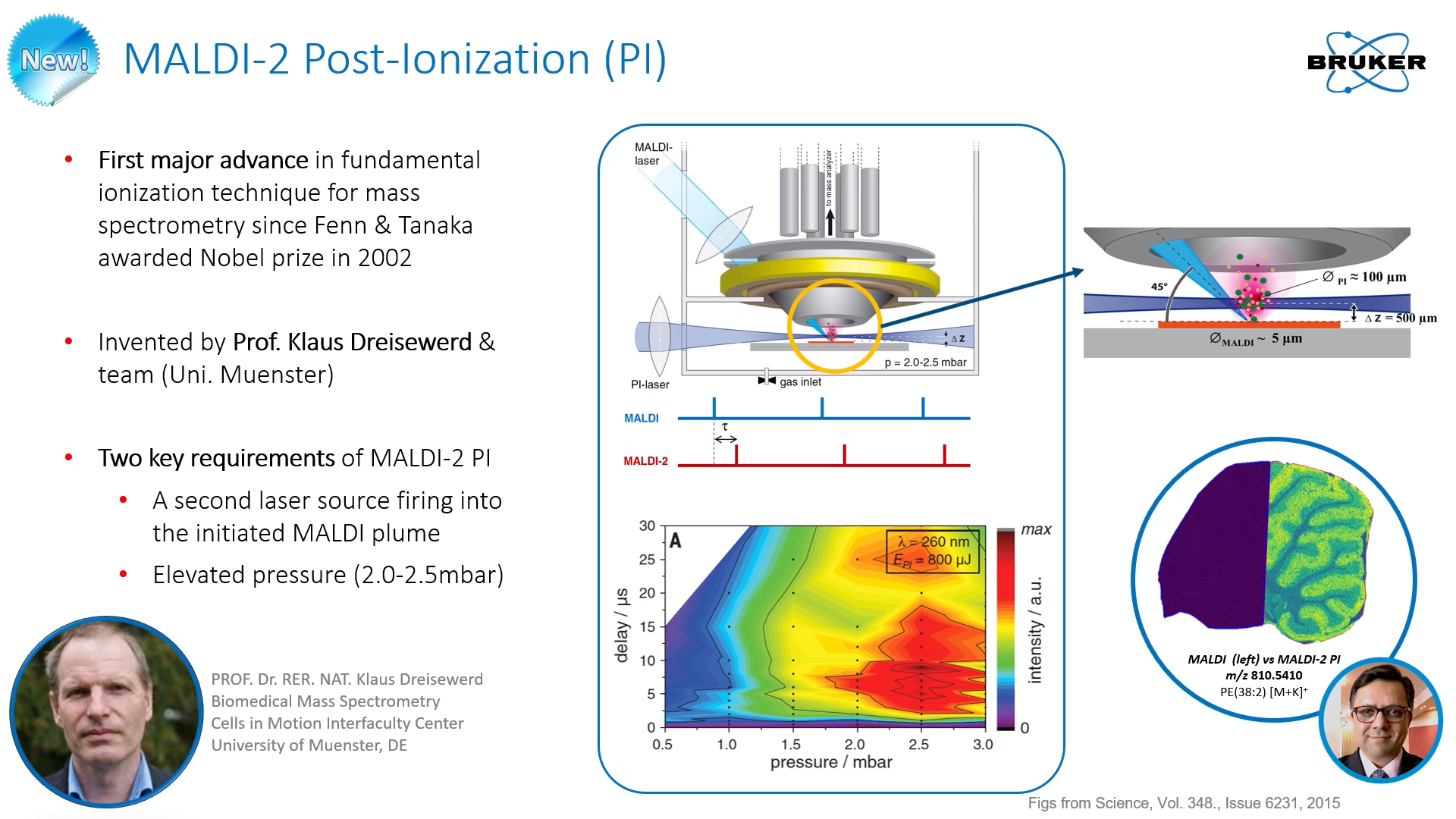
圖 1 Bruker 發表MALDI-2原理:在MALDI源後使用第二道雷射激發中性分子提升電離效率
2015年,德國University of Muenster的Klaus Dreisewerd在《Science》上發表了Post Ionization的MALDI技術,使用第二道雷射激發MALDI一次激發出的離子束,同時配合冷卻氣體(最適化在2.0-2.5 mbar氣壓範圍),距離樣本約5 mm左右,實現了MALDI後的二次離子化。今年Bruker首先完成這項技術商品化,推出安裝在timsTOF fleX系統上的MALDI-2離子源,大幅提升小分子化合物的靈敏度,高達1-3個數量級,甚至有些在原有MALDI中沒有訊號的小分子(如雌二醇estradiol、犬尿氨酸kynurenine、脂溶性維生素)也能穩定地獲得訊號,如圖2所示。
MALDI-2離子源研究先驅,德國University of Muenster生物醫學質譜教授Klaus Dreisewerd表示:「在過去的35年中,MALDI已成為適用於多種應用、獨特而快速的分析工具。現在我們開發的MALDI-2離子源,可以顯著擴展MALDI的應用領域,例如為小分子分析提供更高的靈敏度,以及讓那些無法在傳統MALDI離子源離子化的分子產生訊號。」隨著MALDI成像和SpatialOMx在藥物開發中針對用於組織模型分析價值的增長,研究者追求更高的靈敏度與多功能性,憑藉MALDI-2顯著提高的靈敏度與可應用化合物類型的增加,MALDI-2離子源可以進一步增強質譜的非靶向組織分析。Bruker現在還為其MetaboScape®代謝體學分析軟體提供MALDI-2化合物質譜圖庫。MetaboScape軟體在SCiLS™ Lab成像軟體內可提供分析物自動標注(annotation)功能,直接在組織圖像中對許多代謝物、醣聚物和脂質針對全新CCS演算法進行的標注。
MALDI-2離子源研究先驅,德國University of Muenster生物醫學質譜教授Klaus Dreisewerd表示:「在過去的35年中,MALDI已成為適用於多種應用、獨特而快速的分析工具。現在我們開發的MALDI-2離子源,可以顯著擴展MALDI的應用領域,例如為小分子分析提供更高的靈敏度,以及讓那些無法在傳統MALDI離子源離子化的分子產生訊號。」隨著MALDI成像和SpatialOMx在藥物開發中針對用於組織模型分析價值的增長,研究者追求更高的靈敏度與多功能性,憑藉MALDI-2顯著提高的靈敏度與可應用化合物類型的增加,MALDI-2離子源可以進一步增強質譜的非靶向組織分析。Bruker現在還為其MetaboScape®代謝體學分析軟體提供MALDI-2化合物質譜圖庫。MetaboScape軟體在SCiLS™ Lab成像軟體內可提供分析物自動標注(annotation)功能,直接在組織圖像中對許多代謝物、醣聚物和脂質針對全新CCS演算法進行的標注。
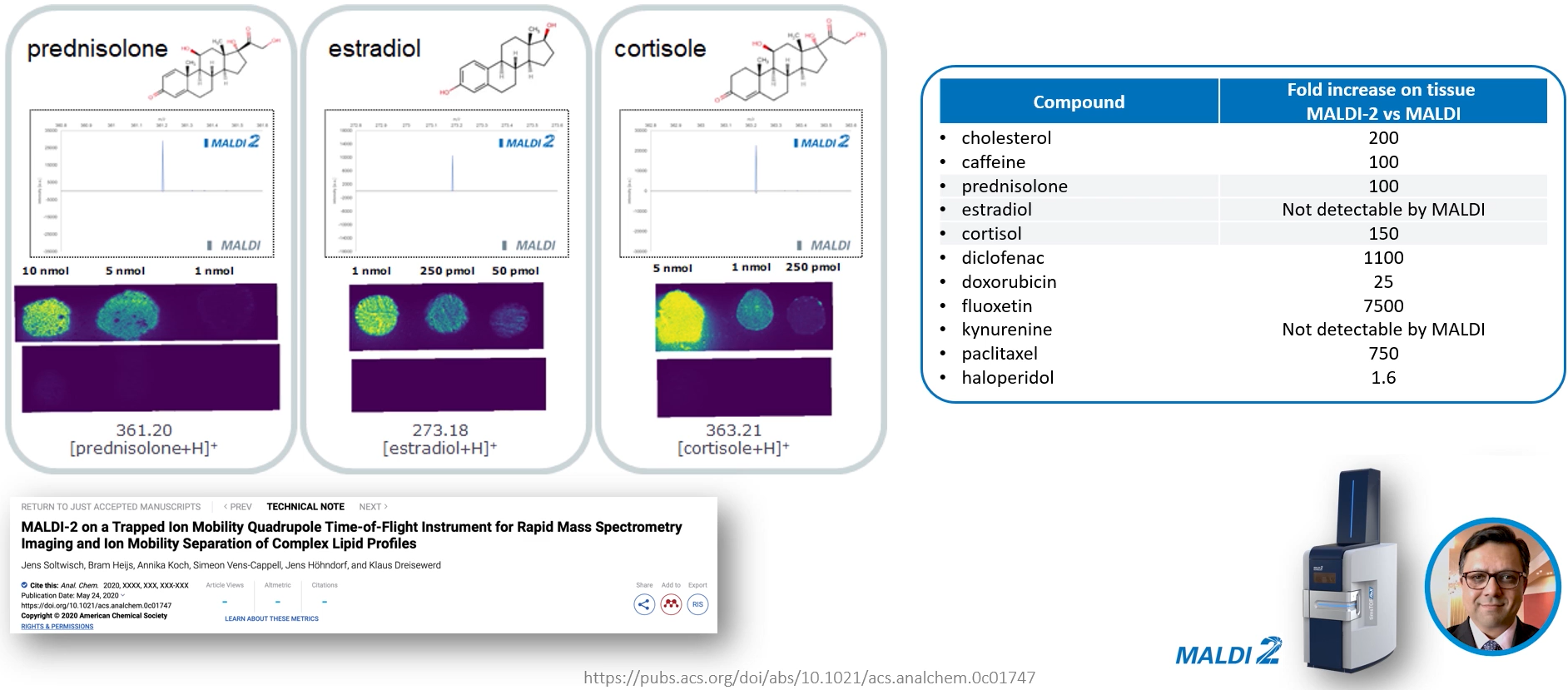
圖 2 使用MALDI-2後,小分子化合物的靈敏度得到數量級的提高
timsTOF fleX在timsTOF Pro基礎上增加了MALDI-2功能,用timsTOF fleX一台儀器即可連接體液、細胞、組織所有研究內容,即空間定位體學(SpatialOMx™)。以前在體學和疾病間總有一道鴻溝,有些研究者做高品質的蛋白質體學、脂質體學或代謝體學等,特別是當他們使用timsTOF Pro時,他們希望和疾病、癌症患者的醫生交談,但這是一個艱難的對話,因為醫生讀的是組織切片。如果是一個習慣於整天看組織切片的人,旁邊需要有人研磨組織,然後執行LC-MS分析。因此兩者之間有一個中斷點,即使體學獲得了豐富的化學資訊,但卻與組織病理的條件失去直接的關聯性。而timsTOF fleX是一台理想的儀器,它構建了一個把體學和疾病兩個領域結合在一起的系統,搭配今年推出的MALDI-2,在m/z之外有離子遷移率的分離,除了得到更豐富的分析資訊,更能對相同質量分子做更細微的分離,如圖3所示。Bruker是第一家從組織中提取細胞,再進行深層次蛋白質體學、代謝體學、脂質體學的公司,在此領域沒有競爭者,如果觀察一個病理組織切片,癌細胞擴散了,到底在哪裡?用SpatialOMx就可以了解,timsTOF fleX,在體學基礎和組織學基礎上,可以看到化學標誌物活動的資訊。
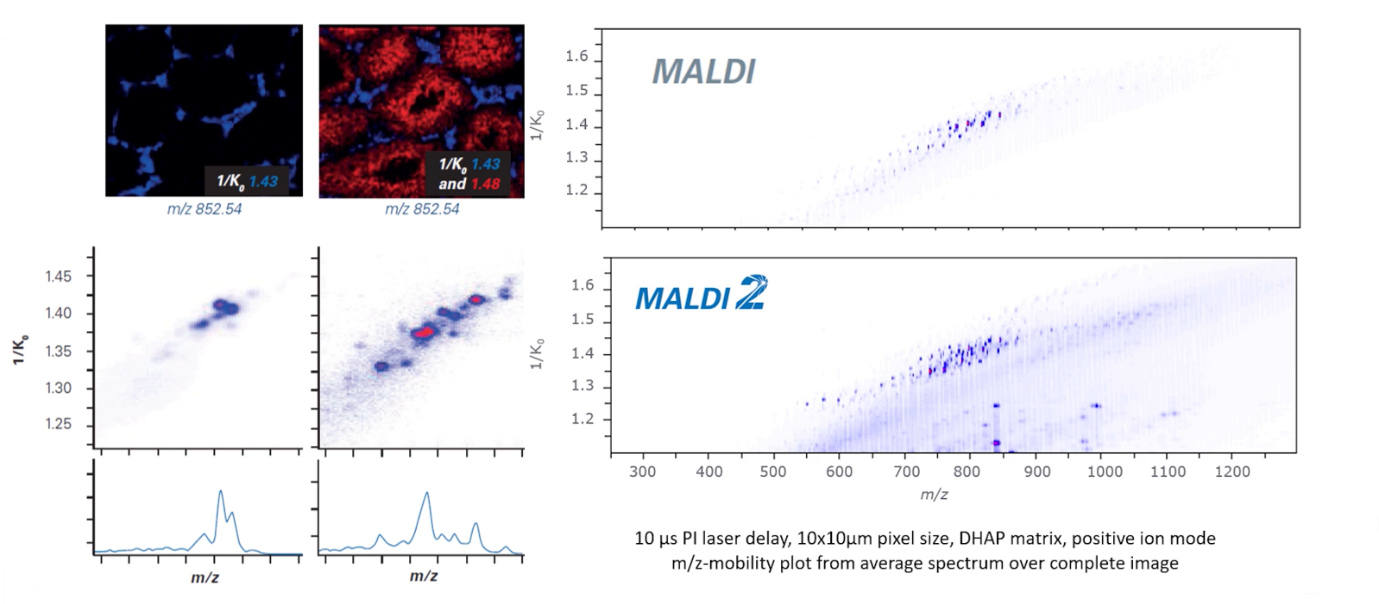
圖 3 在timsTOF fleX上運用MALDI-2後,大幅提升按照CCS離子遷移率分離的化合物訊號和成像品質
新的PRM-PASEF方法讓4D-蛋白質體學分析技術更強大
在上一屆ASMS中,Bruker與Matthias Mann發表了4D蛋白質體的4D-DIA(diaPASEF)技術,並展現其在深度、靈敏度、可靠性方面的優勢,進一步提高30%離子利用率(如圖4),而至本屆ASMS中, diaPASEF技術已臻成熟,進入到實用階段。常用的DIA資料庫搜尋軟體Skyline和Spectrunaut都已經全面支援diaPASEF資料處理,而Spectrunaut在本屆ASMS公佈最新的軟體版本,將功能擴展至4D平臺,進一步提高檢測的效率。
除了大規模的discovery proteomics分析,靶向的targeted proteomics技術同樣在驗證和臨床方面有主要應用前景。2019年發佈的dia-PASEF主要用於快速、大規模的定性,今年ASMS新發佈的PRM-PASEF®主要用於定量,在timsTOF™ Pro上,將PASEF與平行反應監測(PRM)相結合,使其非標記定量(label-free)蛋白質體學技術得到提升,利用TIMS的第4維分離與PASEF的高速掃描優勢,提高了多肽離子的選擇性和靈敏度,增加了標靶離子的數目(圖5),Bruker並與Skyline進行PRM-PASEF方法開發,現在Skyline軟體已經可以分析PRM-PASEF資料並生成定量分析報告。
在上一屆ASMS中,Bruker與Matthias Mann發表了4D蛋白質體的4D-DIA(diaPASEF)技術,並展現其在深度、靈敏度、可靠性方面的優勢,進一步提高30%離子利用率(如圖4),而至本屆ASMS中, diaPASEF技術已臻成熟,進入到實用階段。常用的DIA資料庫搜尋軟體Skyline和Spectrunaut都已經全面支援diaPASEF資料處理,而Spectrunaut在本屆ASMS公佈最新的軟體版本,將功能擴展至4D平臺,進一步提高檢測的效率。
除了大規模的discovery proteomics分析,靶向的targeted proteomics技術同樣在驗證和臨床方面有主要應用前景。2019年發佈的dia-PASEF主要用於快速、大規模的定性,今年ASMS新發佈的PRM-PASEF®主要用於定量,在timsTOF™ Pro上,將PASEF與平行反應監測(PRM)相結合,使其非標記定量(label-free)蛋白質體學技術得到提升,利用TIMS的第4維分離與PASEF的高速掃描優勢,提高了多肽離子的選擇性和靈敏度,增加了標靶離子的數目(圖5),Bruker並與Skyline進行PRM-PASEF方法開發,現在Skyline軟體已經可以分析PRM-PASEF資料並生成定量分析報告。
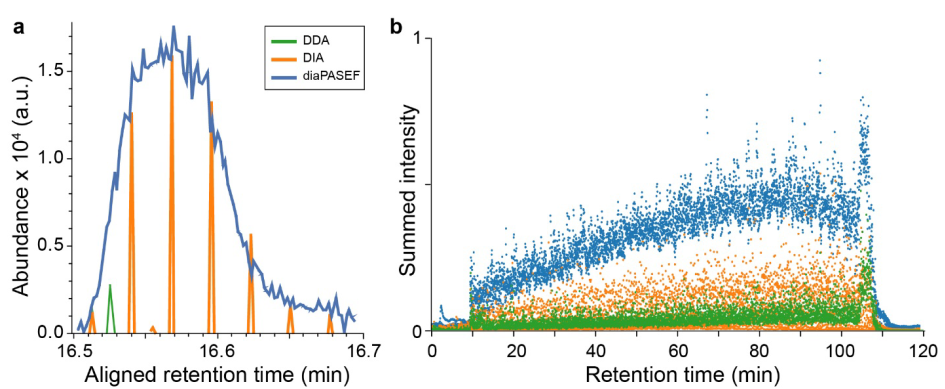
圖 4 不同資料獲取方法的效率。a. 碎片離子的提取離子層析圖,由BSA酶解的雙電荷DLGEEHFK母離子產生,對比DDA、DIA和100% 週期利用的diaPASEF方法;b. 合計的提取離子層析圖,由一次實驗中Hela酶解的多電荷母離子產生,對比DDA、DIA和25% 週期利用的diaPASEF方法
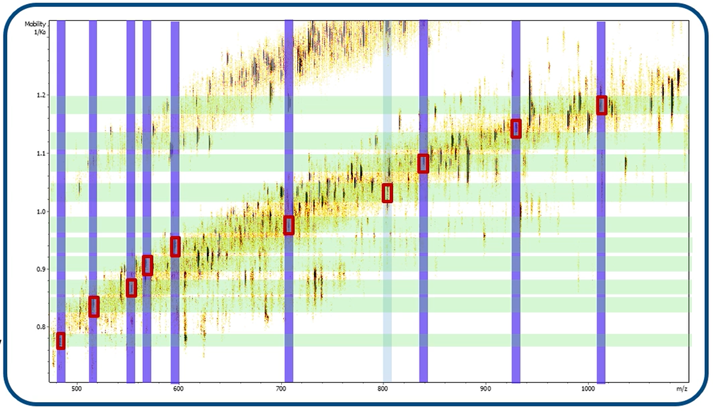
圖 5 PRM-PASEF 在m/z與離子遷移率上選擇(圖中紅框),增加了標靶離子的數目,提高多肽離子的選擇性
哈佛醫學院Dana-Farber Cancer Institute中Brigham and Women's Hospital副教授賈Jarrod Marto博士表示:「自從開始與Bruker合作開發PRM-PASEF以來,我們獲得了大幅進展。timsTOF Pro超快的採集速度和離子遷移率資訊的獨特結合,使我們在臨床大量樣本研究中,能夠可靠地定量分析潛在的候選生物標誌物。此外,利用PRM-PASEF即時調整採集參數功能,增加用戶分析彈性,更可進一步提高離子利用率和分析通量。」
圖6顯示PRM-PASEF的定量表現。當不使用tims時,無論高濃度(125 amol)或低濃度(31.25 amol),碎片沖提的時間有所跳動,並且有許多干擾;然而,當使用tims增加分離維度時,高或低濃度的碎片沖提時間都被聚焦,也免除干擾訊號,此結果展示了tims在PRM-PASEF定量應用的可靠性。
來自Luxembourg Institute of Health的Dr. Antoine Lesur,在今年ASMS中展示了使用timsTOF Pro的PRM-PASEF方法,使用30分鐘梯度在100ng HeLa裂解物基質中,外添加215個目標物進行定量分析,濃度範圍覆蓋5 amol-50 fmol,有良好的檢量線(R2>0.999),如圖7。
圖6顯示PRM-PASEF的定量表現。當不使用tims時,無論高濃度(125 amol)或低濃度(31.25 amol),碎片沖提的時間有所跳動,並且有許多干擾;然而,當使用tims增加分離維度時,高或低濃度的碎片沖提時間都被聚焦,也免除干擾訊號,此結果展示了tims在PRM-PASEF定量應用的可靠性。
來自Luxembourg Institute of Health的Dr. Antoine Lesur,在今年ASMS中展示了使用timsTOF Pro的PRM-PASEF方法,使用30分鐘梯度在100ng HeLa裂解物基質中,外添加215個目標物進行定量分析,濃度範圍覆蓋5 amol-50 fmol,有良好的檢量線(R2>0.999),如圖7。
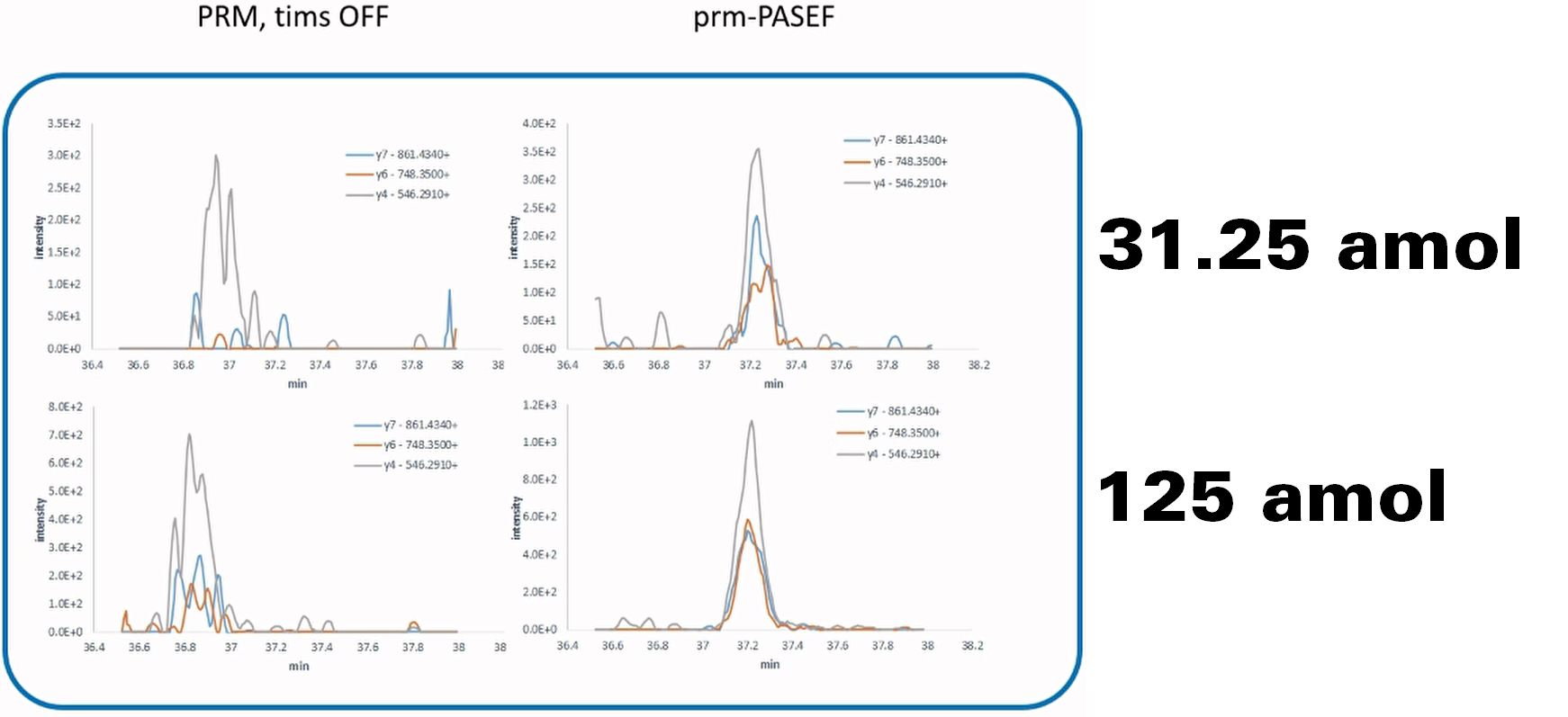
圖 6 PRM-PASEF定量分析效能。左側僅運用四極桿選擇母離子,不同碎片彼此仍存在干擾,且沖提時間跳動;右側選擇離子遷移率後則會消除干擾,並聚焦碎片沖提時間,因此提升定量的準確度
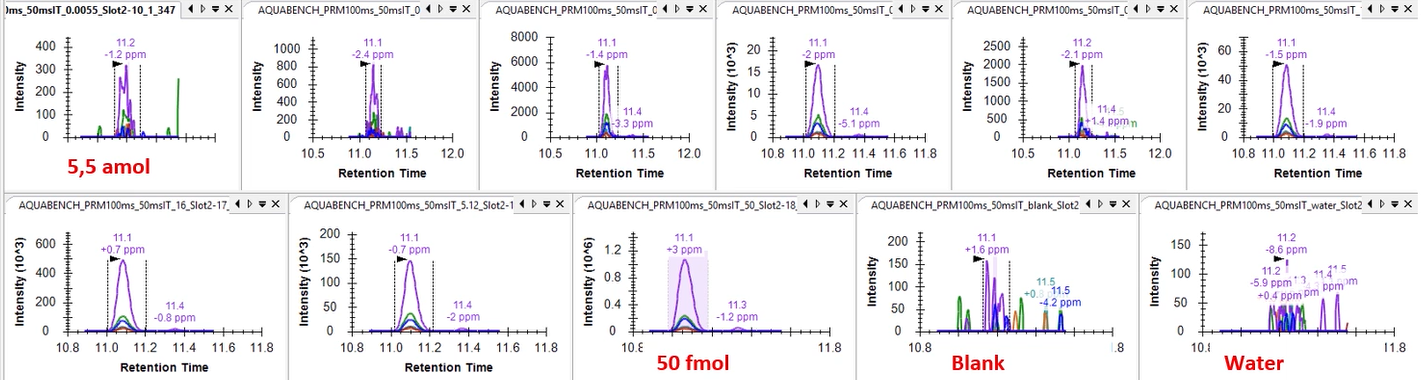
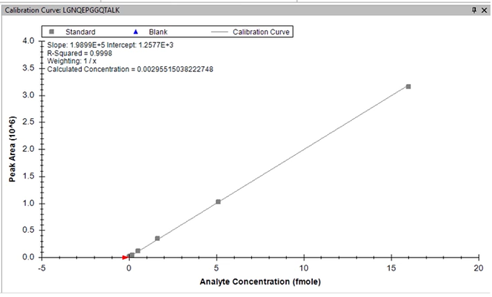
圖 7 基質為100ng HeLa裂解物,同時對215個目標分析物進行定量分析,30分鐘梯度下獲得的該肽段定量結果,濃度範圍覆蓋5 amol-50 fmol,具有良好線性
4D蛋白體學結合深度學習,預測CCS數值提高鑑定可靠性
目前4D蛋白質組中的CCS值可用於相互比對,提高鑑定定性定量的能力。如果能夠提前確定CCS這個數字的絕對常量,那麼將能夠進一步提升其鑑定的可靠性。應用深度學習有望實現這一目標。Matthias Mann在今年ASMS上做“Deep learning the peptide universe from one million peptide collisional cross sections”的口頭報告,展現該領域的最新進展,這為4D蛋白質組技術賦予新的能力。採用獨特的TIMS技術,多肽的CCS值可以被大規模精確測定,最近Matthias Mann教授團隊在bioRxiv上線上發表了題為《Deep learning the collisional cross sections of the peptide universe from a million training samples》的論文,研究中,他們在timsTOF Pro使用PASEF分析了來源於5種生物,並經過預分級的蛋白酶解物,進行大規模肽段碰撞截面積CCS測量。一共採集360針樣本得到570,000 個CCS值,用於深度學習中的模型建立,藉此進行大規模多肽CCS值的預測(圖8)。
目前4D蛋白質組中的CCS值可用於相互比對,提高鑑定定性定量的能力。如果能夠提前確定CCS這個數字的絕對常量,那麼將能夠進一步提升其鑑定的可靠性。應用深度學習有望實現這一目標。Matthias Mann在今年ASMS上做“Deep learning the peptide universe from one million peptide collisional cross sections”的口頭報告,展現該領域的最新進展,這為4D蛋白質組技術賦予新的能力。採用獨特的TIMS技術,多肽的CCS值可以被大規模精確測定,最近Matthias Mann教授團隊在bioRxiv上線上發表了題為《Deep learning the collisional cross sections of the peptide universe from a million training samples》的論文,研究中,他們在timsTOF Pro使用PASEF分析了來源於5種生物,並經過預分級的蛋白酶解物,進行大規模肽段碰撞截面積CCS測量。一共採集360針樣本得到570,000 個CCS值,用於深度學習中的模型建立,藉此進行大規模多肽CCS值的預測(圖8)。
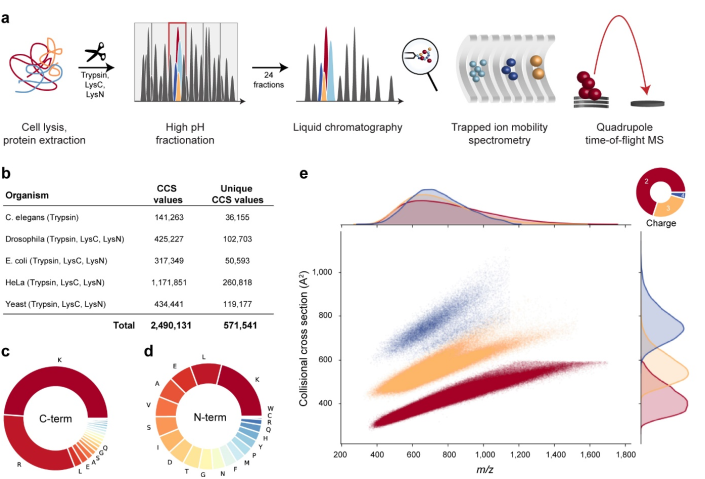
圖 8 a. 從全細胞蛋白質體提取到裂解,分級分離和層析分離至timsTOF質譜儀以PASEF模式運行的工作流程; b. 本研究中按生物源分列的CCS數據組; c. C-terminal胺基酸頻率; d. N-terminal胺基酸頻率; e. 559,979特徵數據點對CCS vs. m/z分布,包括序列修飾與電荷數(以顏色區分),m/z與CCS值的密度分布各別投影在數軸的頂端與右側。
“Run & Done”即時搜尋引擎用於高通量4D-蛋白質體學
蛋白質體資料量大,其資料庫搜尋的時間和計算資源都是導致通量無法提升的要因,在4D蛋白質體中,由於增加了新的維度,其資料計算的消耗資源相比傳統技術又會更加龐大。當前,由於演算法和電腦性能的提升,Real-time的即時搜尋方法成為了新的趨勢,於樣品分析進行數據採集的同時、同步進行數據處理資料庫搜尋的方式能夠大大提升蛋白質組技術的便捷性和檢測通量。
Bruker在ASMS發表,傑出貢獻獎得主John Yates開發的ProLuCID搜尋工具,基於GPU計算的Proteomic Pipeline(IP2)引擎已經可應用於timsTOF Pro產生的4D蛋白質體資料。這個運用GPU運算的獨特軟體由Robin Park博士開發,它允許使用者在資料擷取期間即時搜尋timsTOF Pro的分析資料,資料擷取完成同時即可得到搜尋結果,進行大量多執行緒的平行計算,並將搜尋結果回傳至資料擷取控制軟體,指導質譜資料擷取,從而大幅縮短搜尋等待時間。
蛋白質體資料量大,其資料庫搜尋的時間和計算資源都是導致通量無法提升的要因,在4D蛋白質體中,由於增加了新的維度,其資料計算的消耗資源相比傳統技術又會更加龐大。當前,由於演算法和電腦性能的提升,Real-time的即時搜尋方法成為了新的趨勢,於樣品分析進行數據採集的同時、同步進行數據處理資料庫搜尋的方式能夠大大提升蛋白質組技術的便捷性和檢測通量。
Bruker在ASMS發表,傑出貢獻獎得主John Yates開發的ProLuCID搜尋工具,基於GPU計算的Proteomic Pipeline(IP2)引擎已經可應用於timsTOF Pro產生的4D蛋白質體資料。這個運用GPU運算的獨特軟體由Robin Park博士開發,它允許使用者在資料擷取期間即時搜尋timsTOF Pro的分析資料,資料擷取完成同時即可得到搜尋結果,進行大量多執行緒的平行計算,並將搜尋結果回傳至資料擷取控制軟體,指導質譜資料擷取,從而大幅縮短搜尋等待時間。
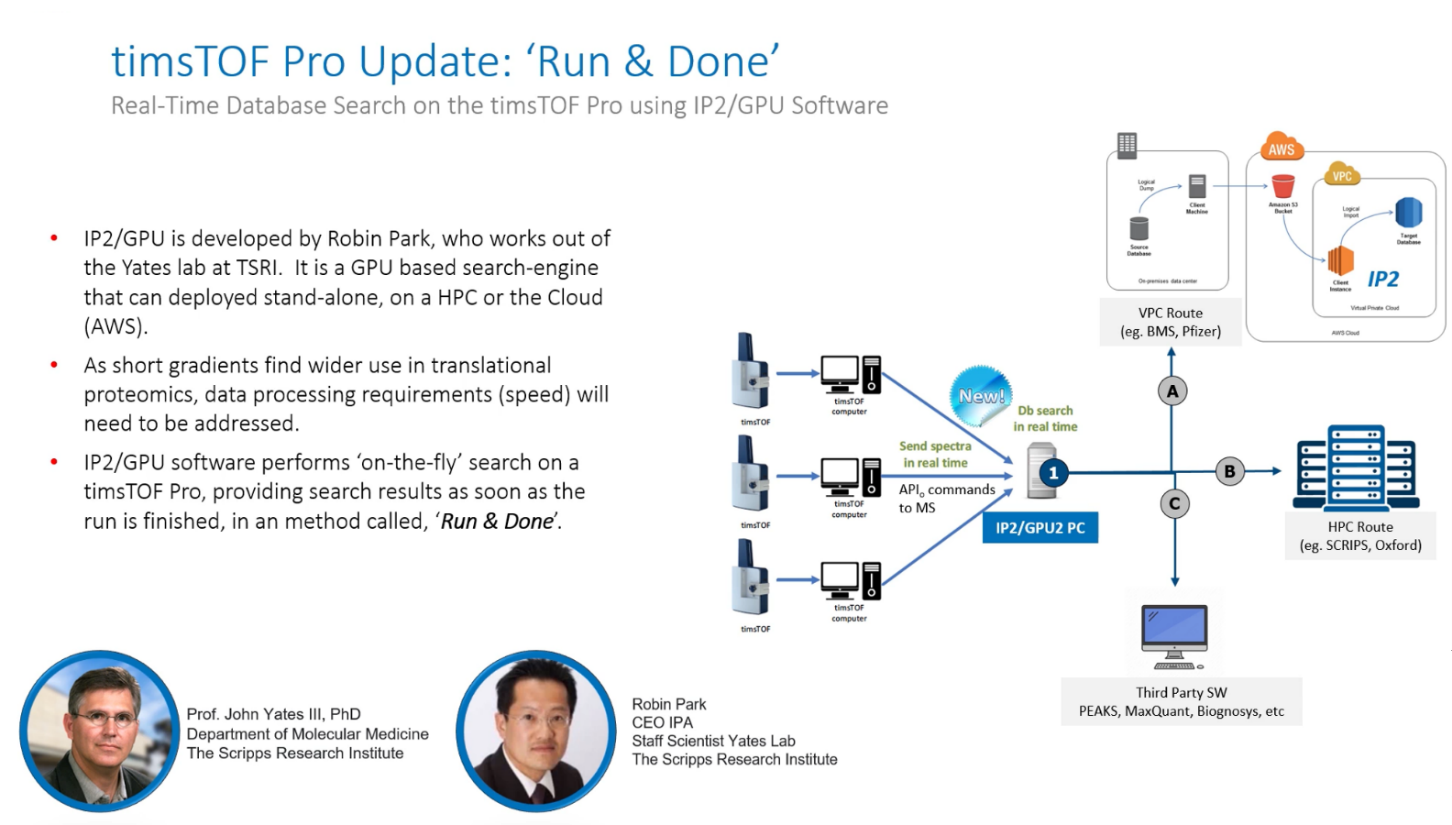
圖 9 timsTOF Pro “Run & Done” 即時搜尋示意圖,一旦完成資料分析也同時完成資料庫搜尋
結語
近三年Bruker質譜針對複雜科研市場推出大量高端產品,顯見重要的三大發展方向是:多體學、製藥(尤其是生物製藥)、空間定位成像技術。這三大領域也是本屆ASMS Bruker的重點。奠基於PASEF的穩定性、靈敏度和離子利用率近100% 的短梯度方法,再隨著MALDI-2、prm-PASEF的推出、dia-PASEF的日益成功,使timsTOF Pro或是timsTOF fleX具有將4D-蛋白質體學轉化至臨床應用的能力。此外,tims獨特的MOMA(Mobility Offset Mass Aligned)特性,能夠區分共同沖提出來的同分異構離子,得到對未知物更加專屬的MS/MS結果。MOMA功能有助於提高使用短梯度時蛋白覆蓋率深度,這對蛋白質體學研究的用戶來說至關重要,如此一來能讓用戶每天在timsTOF Pro上分析>50個樣本。
近三年Bruker質譜針對複雜科研市場推出大量高端產品,顯見重要的三大發展方向是:多體學、製藥(尤其是生物製藥)、空間定位成像技術。這三大領域也是本屆ASMS Bruker的重點。奠基於PASEF的穩定性、靈敏度和離子利用率近100% 的短梯度方法,再隨著MALDI-2、prm-PASEF的推出、dia-PASEF的日益成功,使timsTOF Pro或是timsTOF fleX具有將4D-蛋白質體學轉化至臨床應用的能力。此外,tims獨特的MOMA(Mobility Offset Mass Aligned)特性,能夠區分共同沖提出來的同分異構離子,得到對未知物更加專屬的MS/MS結果。MOMA功能有助於提高使用短梯度時蛋白覆蓋率深度,這對蛋白質體學研究的用戶來說至關重要,如此一來能讓用戶每天在timsTOF Pro上分析>50個樣本。
參考文獻:
- Jens Soltwisch, Hans Kettling, Simeon Vens-Cappell, Marcel Wiegelmann, Johannes Müthing, Klaus Dreisewerd. Mass spectrometry imaging with laser induced postionization. Science 10 Apr 2015: Vol. 348, Issue 6231, pp. 211-215.
- Florian Meier, Andreas-David Brunner, Max Frank, Annie Ha, Eugenia Voytik, Stephanie Kaspar-Schoenefeld, Markus Lubeck, Oliver Raether, Ruedi Aebersold, Ben C. Collins, Hannes L. Röst, Matthias Mann, Parallel accumulation – serial fragmentation combined with data-independent acquisition (diaPASEF): Bottom-up proteomics with near optimal ion usage. bioRxiv preprint first posted online May. 31, 2019.
- Jarrod J Sandow, Giuseppe Infusini, Laura F Dagley, Rune Larsen, Andrew I Webb, Simplified high-throughput methods for deep proteome analysis on the timsTOF Pro. bioRxiv preprint posted online June 3, 2019.
- Florian Meier1, Niklas D. Köhler, Andreas-David Brunner, Jean-Marc H. Wanka, Eugenia Voytik, Maximilian T. Strauss, Fabian J. Theis and Matthias Mann, Deep learning the collisional cross sections of the peptide universe from a million training samples. bioRxiv preprint posted online May 21, 2020.